| ||||||||||||||||||||
|
|||||||||||||||||||||||
J. Clin. Invest.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Measurement of anti-AChR antibodies.
Anti-AChR was measured by conventional radioimmunoassay using 125I- -BuTx (> 200 mCi/mmol, Amersham International, Little Chalfont, UK) to label
human muscle AChR extracted from a mixture of the ischemic muscles of amputated legs (11). The AChR preparation contained mostly
fetal (denervated) type AChR (as shown by reactivity with monoclonal antibodies specific for fetal AChR [12]) with a small amount of
adult-type AChR. 50 µl of prelabeled muscle extract was incubated
with serum dilutions for 2 h at room temperature, and complexes precipitated with excess goat anti-human Ig using normal human serum
as carrier if necessary. Results were expressed as nanomoles of -BuTx
binding sites precipitated per liter of serum, using data from the linear part of the binding curve.
-BuTx (> 200 mCi/mmol, Amersham International, Little Chalfont, UK) to label
human muscle AChR extracted from a mixture of the ischemic muscles of amputated legs (11). The AChR preparation contained mostly
fetal (denervated) type AChR (as shown by reactivity with monoclonal antibodies specific for fetal AChR [12]) with a small amount of
adult-type AChR. 50 µl of prelabeled muscle extract was incubated
with serum dilutions for 2 h at room temperature, and complexes precipitated with excess goat anti-human Ig using normal human serum
as carrier if necessary. Results were expressed as nanomoles of -BuTx
binding sites precipitated per liter of serum, using data from the linear part of the binding curve.
Inhibition of 125I--BuTx binding.
Antibodies directed towards
the 125I--BuTx binding sites were measured by preincubating 50 µl
of the muscle extract with different dilutions of the patient's sera in
50 µl of PTX buffer for 2 h at room temperature before adding 125I--
BuTx at saturating concentrations (2-3 nM). 125I--BuTx-AChR
complexes were precipitated as above by addition of 1 µl of a MG serum pool, to bind AChR, followed by excess goat anti-human IgG.
Results were expressed as nanomoles of binding sites inhibited/liter
of serum, based on the amount of 125I--BuTx binding inhibited by
0.16 µl of each AMC-M serum.
22Na+ flux assay.
AChR function in TE671 cells was measured as
previously described (13, 14). TE671 cells (here called TE671- ) that
express fetal AChR, or TE671-
) that
express fetal AChR, or TE671- , a subline in which over 70% of the
AChR is the adult form (15), were grown under standard conditions and plated at 1-2 � 106 cells per 32 mm well. 2 d later the medium
was removed and replaced with serum or IgG dilutions in Hepes
Locke buffer for 1 h at room temperature. The cells were briefly
washed and the 22Na+ flux measured using 1 min application of 0.2-
0.5 mM carbachol as agonist and 300,000 cpm/ml of 22Na+ in Hepes
Locke buffer. The wells were washed rapidly four times with 2 ml of
Hepes Locke buffer, and the retained 22Na+ counted on a gamma
counter. Results were compared with wells incubated in buffer alone,
after subtraction of cpm in wells pretreated with excess -BuTx to inhibit AChR function. In some experiments serum was tested similarly
after a 30-min preincubation in mAbs to AChR (ascites diluted at
1:250-1:500; [12]). Based on preliminary experiments, this concentration was sufficient to bind all the surface AChR.
, a subline in which over 70% of the
AChR is the adult form (15), were grown under standard conditions and plated at 1-2 � 106 cells per 32 mm well. 2 d later the medium
was removed and replaced with serum or IgG dilutions in Hepes
Locke buffer for 1 h at room temperature. The cells were briefly
washed and the 22Na+ flux measured using 1 min application of 0.2-
0.5 mM carbachol as agonist and 300,000 cpm/ml of 22Na+ in Hepes
Locke buffer. The wells were washed rapidly four times with 2 ml of
Hepes Locke buffer, and the retained 22Na+ counted on a gamma
counter. Results were compared with wells incubated in buffer alone,
after subtraction of cpm in wells pretreated with excess -BuTx to inhibit AChR function. In some experiments serum was tested similarly
after a 30-min preincubation in mAbs to AChR (ascites diluted at
1:250-1:500; [12]). Based on preliminary experiments, this concentration was sufficient to bind all the surface AChR.
Effects of IgG on fetal and adult AChR expressed in Xenopus oocytes.
IgG was purified using protein A sepharose under standard
conditions using a Pierce kit. IgG fractions were dialyzed overnight against Ringer's solution. Expression of human AChR in Xenopus oocytes and measurement of ACh-induced ion currents was performed as described (8, 16). Briefly, oocytes were injected with cRNAs
encoding either fetal (2
 ) or adult (2) type human AChR
subunits. ACh-induced currents were measured by conventional two
microelectrode voltage clamp 2-7 d following injection. Currents evoked by 1 mM ACh were measured before and immediately after a
40-min incubation in IgG at 1:50 dilution.
) or adult (2) type human AChR
subunits. ACh-induced currents were measured by conventional two
microelectrode voltage clamp 2-7 d following injection. Currents evoked by 1 mM ACh were measured before and immediately after a
40-min incubation in IgG at 1:50 dilution.
Results 
We first examined
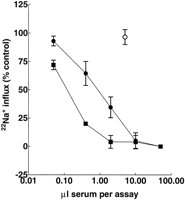
serum anti-AChR antibodies by immunoprecipitation. All
AMC-M sera precipitated 125I--BuTx-AChR at high dilution
(Fig. 1 A). In titrations of sera from AMC-M1, AMC-M2, and
AMC-M4, however, only about 40-50% of the 125I--BuTx
was precipitated even with 4 µl of each serum, compared with
that precipitated when AMC-M3 and AMC-M5 sera (Fig. 1
A), or a serum pool from typical MG patients (not shown)
were tested. This suggested, from previous evidence (17), that
these three AMC-M sera were partially displacing 125I--BuTx
from its binding sites on the AChR. We therefore looked directly for antibodies against the -BuTx binding sites. When AChR was preincubated with the sera before addition of 125I--BuTx, all five AMC-M sera showed strong inhibition of 125I--BuTx binding, with 40-60% inhibition at 4 µl (Fig. 1 B; Table II). Because the AChR in this assay was from denervated
human muscle that expresses mainly fetal AChR (see Methods), these results suggested that each of the AMC-M sera
contained antibodies that bind to the 125I--BuTx site(s) on fetal-type AChR.
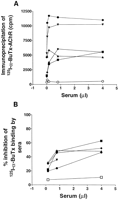
 ), AMC-M2 (
), AMC-M2 ( ), and AMC-M4 (
), and AMC-M4 ( ) precipitate only about 40-50% of the
available 125I--BuTx binding sites, in contrast to AMC-M3 (
) precipitate only about 40-50% of the
available 125I--BuTx binding sites, in contrast to AMC-M3 ( ) and
AMC-M5 (
) and
AMC-M5 ( ) that precipitate all available binding sites. (B) Inhibition of 125I--BuTx binding to ischemic leg muscle AChR by preincubating in AMC-M sera. All the sera strongly inhibit binding of 125I-
-BuTx. An MG serum pool has little effect (
) that precipitate all available binding sites. (B) Inhibition of 125I--BuTx binding to ischemic leg muscle AChR by preincubating in AMC-M sera. All the sera strongly inhibit binding of 125I-
-BuTx. An MG serum pool has little effect ( ). Some of the data
points have been omitted for clarity.
). Some of the data
points have been omitted for clarity.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
To determine whether the AMC-M sera could inhibit fetal AChR function, we
investigated their effect (1:100 dilution) on agonist-induced 22Na+ flux. In each case, AMC-M sera inhibited the 22Na+ flux
into TE671- cells (that express only fetal-type AChR) by > 85% (Table II), as shown in Fig. 2 for AMC-M1 and AMC-M2. 10 healthy control sera at the same dilution had no effect
compared with cells treated with medium alone.
cells (that express fetal-type AChR) by AMC-M sera. AMC-M1 ()
and AMC-M2 () markedly inhibit flux compared with mean results
(±SD) from 10 healthy control sera at 1:100 dilution. The total volume in each assay was 500 µl. Results expressed as% (mean±SD,
n = 3) of control wells in Hepes Locke solution only.
To see whether the inhibitory effects of AMC sera were
confined to fetal AChR, we tested their effects on TE671-
cells that express predominantly adult AChR (15). None of the
AMC-M sera, at 1:100 or 1:25 dilution, reduced AChR function in TE671- cells by more than 20% (Table II).
IgG from AMC-M1 and AMC-M2 was purified by affinity
chromatography. The IgG fractions contained more than 90%
of the anti-AChR antibody and of the activity that inhibited
22Na+ flux (not shown). AMC-M2 IgG at 1:50 dilution markedly reduced ACh-induced currents in Xenopus oocytes that
had been injected with cRNA for the subunits of human fetal
AChR (2), but had no effect on Xenopus oocytes injected
with the subunits for human adult AChR (2) (Fig. 3).
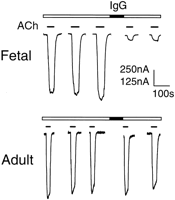
2) but not in those expressing adult type (2). The scale bar represents 250 nA for fetal AChR and 125 nA for adult AChR. Similar results were previously reported with IgG from AMC-M1 (8).
Competition with anti-AChR monoclonal antibodies.
The
specificity of the sera for determinants on fetal-type AChR
was further confirmed by the ability of fetal-specific mAbs to
protect the AChR from the inhibitory actions that AMC-M1
and AMC-M2 sera exerted on 22Na+ flux. Three anti-AChR
mAbs (B8, C2, and C9) that are fetal AChR-specific and bind
to the subunit (L. Jacobson, A. Vincent, and D. Beeson. 1996. In preparation), protected the AChR from subsequent
inhibition by AMC-M1 and AMC-M2 (Fig. 4). By contrast,
mAb B3 that binds to the subunit on fetal and adult AChR
(Jacobson, 1996) did not protect AChR function.

from inhibition by AMC-M1 and AMC-M2 serum by mAbs to fetal-AChR. TE671 cells were preincubated with the mAbs (1:250-1:500)
before washing and addition of AMC-M1 (1:1000) or AMC-M2
(1:250) for 30 min. 22Na+ flux results were expressed as %
(mean±SD, n = 3) of Hepes Locke treated controls.
The subunit specificity of the mAbs is indicated in the legends.
Anti-AChR in serum from unselected AMC mothers.
AMC-M1 and AMC-M5 had no symptoms or signs of MG, in
spite of raised levels of anti-AChR. We therefore assayed 20 sera from further mothers who had an obstetric history of
AMC of unknown cause. The sera were negative for anti-AChR antibodies by immunoprecipitation and did not inhibit
125I--BuTx binding to fetal-type AChR (not shown). However, three of these sera inhibited fetal AChR function in the
22Na+ flux assay by more than 20%, suggesting that there could
be serum antibodies or other factors involved in these cases.
Only 1 of 10 anti-AChR positive MG sera inhibited more
strongly (Fig. 5).
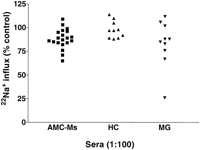
cells by sera from
mothers with obstetric histories of AMC (AMC-Ms) without anti-AChR antibodies. Results are compared with sera from typical MG
patients (MG) and from healthy young controls (HC) on carbachol-induced 22Na+ flux in TE671- cells. Wells were preincubated with
sera at 1:100 dilution. Three AMC-M results (74, 71, and 65%) were
significantly different (P < 0.002, Student's t test; mean of three to
eight determinations) from the mean (98±9, n = 10) of the healthy
control results.
Discussion
We have characterized the serum anti-AChR antibodies from five mothers whose obstetric histories had been complicated by AMC. In AMC-M1 and AMC-M2, recurrent severe intrauterine paralysis was responsible for stillbirths or termination of pregnancy (8, 9). In AMC-M3-5 the babies had both AMC and neonatal MG but survived with treatment (7). Neither AMC-M1 nor AMC-M5 showed symptoms of MG (7). Despite these differing clinical presentations, sera from all five women inhibited fetal AChR function but had little effect on the function of adult AChR. Some inhibition of fetal AChR function was also seen in three of 20 sera from other mothers with histories of fetal AMC. Thus, serum factor(s) that interfere with the function of AChR in the developing fetus may, although occurring rarely, be an important and potentially treatable cause of AMC.
IgG purified from AMC-M1 and AMC-M2 inhibited the
ACh-induced currents of fetal, but not adult, AChR expressed
in Xenopus oocytes (Fig. 3 and reference 8). ACh binds at the
interfaces between the two subunits and their adjacent subunits. These sites differ in antigenicity and affinity for 125I--
BuTx on Torpedo AChR (18). Both fetal and adult forms of AChR have one / site; the second site in fetal AChR is
formed by /, and in adult AChR by / (19). It is possible
that the AMC-M antibodies inhibit fetal AChR function by
binding to the / ACh binding site which is unique to fetal
AChR. The fact that AMC-M sera also strongly inhibited approximately half the 125I--BuTx binding sites on AChR extracted from denervated human muscle (AMC-M1, M2, and
M4; Fig. 1 B), and on surface AChR in TE671- cells (AMC-M1
and AMC-M2, data not shown), is consistent with this hypothesis. Previous reports have found a relationship between inhibition of AChR function and inhibition of 125I--BuTx binding
with some MG sera (20, 21). Although AMC is found in only
about 10% of babies with neonatal MG (7, 22), the inhibition
by serum of 125I--BuTx binding to fetal/denervated AChR
might be helpful in identifying mothers whose fetuses are at
risk.
Preference of MG serum antibodies for fetal/denervated
AChR has been demonstrated in previous studies (e.g. 7, 20, 21, 23, 24). Vernet-der Garabedian et al. (7) recently showed
that mothers who transmit neonatal MG to their babies have a
high proportion of antibodies that, in immunoprecipitation assays, bind to fetal AChR rather than to adult AChR. However,
they did not find this preference in cases of neonatal MG with
AMC (described as fetal damage in their paper), presumably
because they did not look specifically for antibodies that inhibit 125I--BuTx binding to fetal AChR.
The results of this study raise several other questions. AMC-1 and AMC-5 had high anti-AChR levels by immunoprecipitation and yet did not have MG. Antibodies to AChR are usually considered diagnostic for MG, and it is very rare to find high levels of anti-AChR antibody in individuals without any history of weakness (4). Anti-AChR antibodies in MG lead to loss of functional muscle (adult-type) AChR by a variable combination of complement-dependent lysis of the neuromuscular junction, accelerated degradation of AChR and direct block of AChR function (4). It would be interesting to clone IgG antibodies from mothers of AMC babies, using combinatorial expression libraries. One could then test the epitope specificity and pathogenicity of the individual Fabs or reconstructed IgGs.
Another question relates to the antigenic stimulus responsible for inducing the highly specific inhibitory antibodies found in AMC-Ms. Although fetal AChR is present in the normal thymus and some muscles (25, 26), it is possible that the antibodies in AMC occur as an immune response to fetal muscle during pregnancy. It will be interesting to compare the frequency of these antibodies in other MG patients, particularly between parous and nonparous women.
It has been suggested that the susceptibility of extraocular
muscles to myasthenic weakness reflects the presence of fetal AChR on the multiply innervated tonic fibers (27). Although there is some expression of AChR subunit mRNA in extraocular muscle (26), there is more AChR subunit mRNA
(MacLennan, C., D. Beeson, A. Vincent, J. Newsom-Davis,
manuscript submitted for publication), and the lack of any ocular symptoms in AMC-M1 and AMC-M5 argues against antibodies to fetal AChR causing ocular muscle weakness.
The occurrence of high antibody titers against functional
determinants on fetal AChR make it difficult to manage pregnancy in these cases. However, there must be increasing
amounts of adult AChR made throughout the second and
third trimesters since some mRNA for the subunit is expressed by 14 weeks (MacLennan, C., D. Beeson, A. Vincent,
J. Newsom-Davis, in press) and most of the fetal form is lost by
33 wk gestation (28). Therefore treatment might be required
for a limited period during midpregnancy only. Plasma exchange and intravenous immunoglobulin infusions might reduce the level of circulating antibodies to an extent that is compatible with fetal movement. Furthermore, the ability of
mouse monoclonal antibodies to prevent binding of AMC-M1
and -M2 antibodies suggests the possibility of blocking the
binding by monoclonal Fab fragments, or by peptides representing the fetal-specific functional epitope on the AChR. Alternatively, it may be possible to block the active Fc-mediated
transport of maternal antibody across the placenta for a limited period of time during which the fetus is most at risk.
The causes of AMC are multifactorial: some are clearly genetic, but others are unknown. Environmental factors, viral infections, and maternal neurological or muscle disease associated with reduced fetal movements, have been implicated (1). When there is a recurrence in the absence of maternal disease, the etiology is usually presumed to be an autosomal recessive gene. We have shown that maternal antibodies are the cause in some cases. In addition to the five cases characterized here, we have screened sera from 20 mothers with an obstetric history of AMC; none were positive for anti-AChR but three of the sera inhibited AChR function at 1:100 dilution, albeit by a modest amount. Cases of neonatal MG with undetectable anti-AChR have been reported, and seronegative MG appears to be due to antibodies binding to other muscle determinants that indirectly affect AChR function (29). It is possible that maternal antibodies directed at other determinants on fetal muscle may cause muscle weakness or damage in some anti-AChR negative AMC cases. Alternatively, circulating non-Ig inhibitory factors may be present.
The role of maternally derived antibodies in fetal damage is not limited to muscle. In particular, anti-Ro antibodies, from both symptomatic and asymptomatic mothers, can cause congenital heart block (30). Placental transfer of human anti-nuclear antigen antibodies has been demonstrated in mice injected with IgG from a patient with systemic lupus erythematosus (31). This mouse model should be employed to investigate the possible role of antibodies to other fetal specific antigens, or to neuronal antigens that are exposed during fetal development, in causing fetal death or congenital abnormalities.
Footnotes
Address correspondence to Dr. Angela Vincent, Neurosciences Group, Institute of Molecular Medicine, John Radcliffe Hospital, Oxford OX3 9DU. Phone: 44-1865-222323; FAX: 44-1865-222402; E-mail: neurosciences@imm.ox.ac.uk
Received for publication 5 June 1996 and accepted in revised form 23 September 1996.
1. Abbreviations used in this paper: ACh, acetylcholine; AChR, acetylcholine receptor; AMC, arthrogryposis multiplex congenita; AMC-M, mother of AMC offspring;-BuTx, -bungarotoxin; MG,
myasthenia gravis.
Acknowledgments
We thank the Myasthenia Gravis Association/Muscular Dystrophy Group and the Medical Research Council of Great Britain for support. S. Riemersma was assisted by Biomed 1 project PL93 1100. We are very grateful to Mr. Leslie Jacobson, Dr. Nick Willcox, and Mr. Paul Plested for their help, and to Drs. Susan Huson, Louise Brueton, Jane Hurst, and other clinical geneticists who provided serum samples from their cases.
References
| 1. |
Hall, J.G.
,
S.D. Reed
, and
G. Greene
(1982)
The distal arthrogryposes:
delineation of new entities review and nosologic discussion.
Am. J. Med.
Genet.
11:
185-239
[Medline] review and nosologic discussion.
Am. J. Med.
Genet.
11:
185-239
[Medline]
|
| 2. | Drachman, D.B. , and L. Sokoloff (1966) The role of movement in embryonic joint development. Rev. Biol. 14: 401-420 |
| 3. | Jago, R.N. (1970) Arthrogryposis following treatment of maternal tetanus with muscle relaxants. Arch. Dis. Childhood. 45: 277-279 [Medline] |
| 4. |
Drachman, D.B.
(1994)
Myasthenia gravis.
N. Engl. J. Med.
330:
1797-1810
|
| 5. | Claudio, T. 1989. Molecular genetics of acetylcholine receptor-channels. In Frontiers in Molecular Neurobiology. D.M. Glover, and B.D. Hames, editors. IRL Press, Oxford. 63-142. |
| 6. | Morel, E. , B. Eymard , B. Vernet-der Garabedian , C. Pannier , O. Dulac , and J.-F. Bach (1988) Neonatal myasthenia gravis: a new clinical and immunologic appraisal on 30 cases. Neurology. 38: 138-142 [Abstract] |
| 7. | Vernet-der Garabedian, B. , M. Lacokova , B. Eymard , E. Morel , M. Faltin , J. Zajac , O. Sadovsky , M. Dommergues , P. Tripon , and J.F. Bach (1994) Association of neonatal myasthenia gravis with antibodies against the fetal acetylcholine receptor. J. Clin. Invest. 94: 555-559 [Medline] |
| 8. | Vincent, A. , C. Newland , L. Brueton , D. Beeson , S. Riemersma , S.M. Huson , and J. Newsom-Davis (1995) Arthrogryposis multiplex congenita with maternal autoantibodies specific for a fetal antigen. Lancet. 346: 24-25 [Medline] |
| 9. | Barnes, P.R.J. , D.J. Kanabar , L. Brueton , J. Newsom-Davis , S.M. Huson , N.P. Mann , and D. Hilton-Jones (1995) Recurrent congenital arthrogryposis leading to a diagnosis of myasthenia gravis in an initially asymptomatic mother. Neuromusc. Dis. 5: 59-65 [Medline] |
| 10. | Vincent, A., S. Riemersma, D. Beeson, S. Huson, L. Brueton, C. Newland, and J. Newsom-Davis. 1996. Arthrogryposis multiplex congenita associated with maternal antibodies to fetal acetylcholine receptor. Neurology. 46: A112. |
| 11. | Vincent, A. , and J. Newsom-Davis (1985) Acetylcholine receptor antibody as a diagnostic test for myasthenia gravis: results in 153 validated cases and 2967 diagnostic assays. J. Neurol. Neurosurg. Psychiatry. 48: 1246-1252 [Abstract] |
| 12. | Whiting, P.J. , A. Vincent , M. Schluep , and J. Newsom-Davis (1986) Monoclonal antibodies that distinguish between normal and denervated human acetylcholine receptor. J. Neuroimmunol. 11: 223-235 [Medline] |
| 13. | Lang, B. , G. Richardson , J. Rees , A. Vincent , and J. Newsom-Davis (1988) Plasma from myasthenia gravis patients reduces acetylcholine receptor agonist-induced Na+ flux into TE671 cell line. J. Neuroimmunol. 19: 141-148 [Medline] |
| 14. | Yamamoto, T. , A. Vincent , T.A. Ciulla , B. Lang , I. Johnston , and J. Newsom-Davis (1991) Seronegative myasthenia gravis: A plasma factor inhibiting agonist-induced acetylcholine receptor function copurifies with IgM. Ann. Neurol. 30: 550-557 [Medline] |
| 15. |
Beeson, D.
,
M. Amar
,
I. Bermudez
,
A. Vincent
, and
J. Newsom-Davis
(1996)
Stable functional expression of the adult subtype of human muscle
acetylcholine receptor following transfection of the human
rhabdomyosarcoma cell line TE671 with cDNA encoding the subunit.
Neurosci. Lett.
207:
57-60
[Medline]
|
| 16. | Newland, C.F. , D. Beeson , A. Vincent , and J. Newsom-Davis (1995) Functional and non-functional isoforms of the human muscle acetylcholine receptor. J. Physiol. (Lond.). 489: 767-778 [Abstract] |
| 17. | Lang, B. , A. Vincent , J. Newsom-Davis , and . ( (1982) . Purification of anti-acetylcholine receptor antibody from patients with myasthenia gravis. J. Immunol. Meth. 51: 371-381 [Medline] |
| 18. | Dowding, A.J. , and Z.H. Hall (1987) Monoclonal antibodies specific for each of the two toxin-binding sites of Torpedo acetylcholine receptor. Biochemistry. 26: 6372-6381 [Medline] |
| 19. | Karlin, A. , and M.H. Akabas (1995) Toward a Structural Basis for the Function of Nicotinic Acetylcholine Receptors and Their Cousins. Neuron. 15: 1231-1244 [Medline] |
| 20. | Hall, Z.W. , S. Pizzighella , Y. Gu , S. Vicini , and S.M. Schuetze (1987) Functional inhibition of acetylcholine receptors by antibodies in myasthenic sera. Ann. NY Acad. Sci. 505: 272-285 [Medline] |
| 21. | Burges, J. , D.W. Wray , S. Pizzighella , Z. Hall , and A. Vincent (1990) A myasthenia gravis plasma immunoglobulin reduces miniature endplate potentials at human endplates in vitro. Muscle Nerve. 13: 407-413 [Medline] |
| 22. |
Dinger, J.
, and
B. Prager
(1993)
Arthrogryposis multiplex in a newborn
of a myasthenic mothercase report and literature.
Neuromusc. Dis.
3:
335-339
[Medline]
|
| 23. | Weinberg, C.B. , and Z.W. Hall (1979) Antibodies from patients with myasthenia gravis recognize determinants unique to extrajunctional acetylcholine receptors. Proc. Natl. Acad. Sci. USA. 76: 504-508 [Abstract] |
| 24. | Vincent, A. , and J. Newsom-Davis (1982) Acetylcholine receptor antibody characteristics in myasthenia gravis. I. Patients with generalized myasthenia or disease restricted to ocular muscles. Clin. Exp. Immunol. 49: 257-265 [Medline] |
| 25. | Schluep, M. , N. Willcox , A. Vincent , G.K. Dhoot , and J. Newsom-Davis (1987) Acetylcholine receptors in human thymic myoid cells in situ: an immunohistological study. Ann. Neurol. 22: 212-222 [Medline] |
| 26. | Horton, R.M. , A.A. Manfredi , and B-M. Conti-Tronconi (1993) The "embryonic" gamma subunit of the nicotinic acetylcholine receptor is expressed in adult extraocular muscle. Neurology. 43: 983-985 [Abstract] |
| 27. | Kaminski, H.J. , E. Maas , P. Spiegel , and R.L. Ruff (1990) Why are eye muscles frequently involved in myasthenia gravis? Neurology. 40: 1663-1669 [Medline] |
| 28. | Hesselmans, L. , F. Jennekens , C. van den Oord , H. Veldman , and A. Vincent (1993) Immunoreactivity to the acetylcholine receptor in developing human muscle. Anat. Rec. 236: 553-562 [Medline] |
| 29. | Barrett-Jolley, R. , A. Vincent , N. Byrne , J. Newsom-Davis , and . ( (1994) . Plasma from patients with seronegative myasthenia gravis inhibit nAChR responses in the TE671/RD cell line. Pflueg. Arch. Euro. 428: 492-498 |
| 30. | Olson, N.Y. , and C.B. Lindsley (1987) Neonatal lupus syndrome. Am. J. Dis. Child. 141: 908-910 [Abstract] |
| 31. | Guzman-Enriquez, L. , E. Avalos-Diaz , and R. Herrera-Esparza (1990) Transplacental transfer of human antinuclear antibodies in mice by injection of lupus IgG in pregnant animals. J. Rheumatol. 17: 52-56 [Medline] |
This article has been cited by other articles:
 |
R. Watson, Y. Jiang, I. Bermudez, L. Houlihan, L. Clover, K. McKnight, J. H. Cross, I. K. Hart, A. Roubertie, J. Valmier, Y. Hart, J. Palace, D. Beeson, A. Vincent, and B. Lang Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis Neurology, July 13, 2004; 63(1): 43 - 50. [Abstract] [Full Text] [PDF] |
||||
 |
A. VINCENT, J. McCONVILLE, M. E. FARRUGIA, J. BOWEN, P. PLESTED, T. TANG, A. EVOLI, I. MATTHEWS, G. SIMS, P. DALTON, L. JACOBSON, A. POLIZZI, F. BLAES, B. LANG, D. BEESON, N. WILLCOX, J. NEWSOM-DAVIS, and W. HOCH Antibodies in Myasthenia Gravis and Related Disorders Ann. N.Y. Acad. Sci., September 1, 2003; 998(1): 324 - 335. [Abstract] [Full Text] [PDF] |
||||
|
|
H. SHIONO, I. ROXANIS, W. ZHANG, G. P. SIMS, A. MEAGER, L. W. JACOBSON, J-L. LIU, I. MATTHEWS, Y-L. WONG, M. BONIFATI, K. MICKLEM, D. I. STOTT, J. A. TODD, D. BEESON, A. VINCENT, and N. WILLCOX Scenarios for Autoimmunization of T and B Cells in Myasthenia Gravis Ann. N.Y. Acad. Sci., September 1, 2003; 998(1): 237 - 256. [Abstract] [Full Text] [PDF] |
||||
|
|
L. JACOBSON, A. POLIZZI, and A. VINCENT An Animal Model of Maternal Antibodymediated Arthrogryposis Multiplex Congenita (AMC) Ann. N.Y. Acad. Sci., May 13, 1998; 841(1): 565 - 567. [Full Text] |
||||
|
|
A. VINCENT, L. JACOBSON, P. PLESTED, A. POLIZZI, T. TANG, S. RIEMERSMA, C. NEWLAND, S. GHORAZIAN, J. FARRAR, C. MACLENAN, N. WILLCOX, D. BEESON, and J. NEWSDOM-DAVIS Antibodies Affecting Ion Channel Function in Acquired Neuromyotonia, in Seropositive and Seronegative Myasthenia Gravis, and in Antibody-mediated Arthrogryposis Multiplex Congenita Ann. N.Y. Acad. Sci., May 13, 1998; 841(1): 482 - 496. [Full Text] |
||||
 |
S. Brownlow, R. Webster, R. Croxen, M. Brydson, B. Neville, J.-P. Lin, A. Vincent, J. Newsom-Davis, and D. Beeson Acetylcholine receptor {{delta}} subunit mutations underlie a fast-channel myasthenic syndrome and arthrogryposis multiplex congenita J. Clin. Invest., July 1, 2001; 108(1): 125 - 130. [Abstract] [Full Text] [PDF] |
||||
|
|
A. Al-Shekhlee, N. Robin, H. J. Kaminski, A. Polizzi, M. Ruggieri, and A. Vincent Pyridostigmine-induced microcephaly Neurology, June 12, 2001; 56(11): 1606 - 1607. [Full Text] [PDF] |
||||
 |
A. Vincent, D. Beeson, and B. Lang Molecular targets for autoimmune and genetic disorders of neuromuscular transmission FEBS J., December 1, 2000; 267(23): 6717 - 6728. [Abstract] [Full Text] |
||||
|
|
B. Buchwald, M. de Baets, G.-J. Luijckx, and K. V. Toyka Neonatal Guillain-Barre syndrome: Blocking antibodies transmitted from mother to child Neurology, October 1, 1999; 53(6): 1246 - 1246. [Abstract] [Full Text] |
||||
|
|
A. P. Batocchi, L. Majolini, A. Evoli, M. M. Lino, C. Minisci, and P. Tonali Course and treatment of myasthenia gravis during pregnancy Neurology, February 1, 1999; 52(3): 447 - 447. [Abstract] [Full Text] [PDF] |
||||
|
|
L. Jacobson, A. Polizzi, G. Morriss-Kay, and A. Vincent Plasma from human mothers of fetuses with severe arthrogryposis multiplex congenita causes deformities in mice J. Clin. Invest., April 1, 1999; 103(7): 1031 - 1038. [Abstract] [Full Text] |
||||
| ||||||||||||||||||||||||||||||||||||||||||||
 ,
,  Abstract
Abstract

 7 M.
7 M.